5 Usage (R API)
The following examples illustrate the functionality of {biodosetools}’s R API to perform dose-effect fitting and dose estimation for the dicentric and translocation assays. The equivalent examples using the {shiny} user interface can be found in Chapter 4.
The full {biodosetools} R API reference detailing the available functions and parameters can be found on the project’s website at https://biodosetools-team.github.io/biodosetools/reference/.
5.1 Dicentrics dose-effect fitting
5.1.1 Input count data
The first step is to input the count data. This step is accomplished in R by calling the calculate_aberr_table() function. This will calculate the total number of cells (\(N\)), total number of aberrations (\(X\)), as well as mean (\(\bar{y}\)), variance (\(\sigma^{2}\)), dispersion index (\(\sigma^{2}/\bar{y}\)), and \(u\)-value.
count_data <- system.file("extdata", "count-data-barquinero-1995.csv",
package = "biodosetools"
) |>
utils::read.csv() |>
calculate_aberr_table(type = "count")
count_data## # A tibble: 11 × 13
## D N X C0 C1 C2 C3 C4 C5 mean var DI
## <dbl> <int> <dbl> <int> <int> <int> <int> <int> <int> <dbl> <dbl> <dbl>
## 1 0 5000 8 4992 8 0 0 0 0 0.0016 0.00160 0.999
## 2 0.1 5002 14 4988 14 0 0 0 0 0.00280 0.00279 0.997
## 3 0.25 2008 22 1987 20 1 0 0 0 0.0110 0.0118 1.08
## 4 0.5 2002 55 1947 55 0 0 0 0 0.0275 0.0267 0.973
## 5 0.75 1832 100 1736 92 4 0 0 0 0.0546 0.0560 1.03
## 6 1 1168 109 1064 99 5 0 0 0 0.0933 0.0933 0.999
## 7 1.5 562 100 474 76 12 0 0 0 0.178 0.189 1.06
## 8 2 333 103 251 63 17 2 0 0 0.309 0.353 1.14
## 9 3 193 108 104 72 15 2 0 0 0.560 0.466 0.834
## 10 4 103 103 35 41 21 4 2 0 1 0.882 0.882
## 11 5 59 107 11 19 11 9 6 3 1.81 2.09 1.15
## # ℹ 1 more variable: u <dbl>5.1.2 Perform fitting
To perform the fitting in R we call the fit() function, whilst selecting the appropriate aberr_module ("dicentrics" in this example), model_formula ("lin-quad" or "lin" for LQ or L models, respectively), and model_family ("automatic", "poisson", or "quasipoisson") parameters:
fit_results <- fit(
count_data = count_data,
model_formula = "lin-quad",
model_family = "automatic",
aberr_module = "dicentrics"
)The fit_results object is a list that contains all necessary information about the count data as well as options selected when performing the fitting. This is a vital step to ensure traceability and reproducibility. Below we can see its elements:
names(fit_results)## [1] "fit_raw_data" "fit_formula_raw" "fit_formula_tex"
## [4] "fit_coeffs" "fit_cor_mat" "fit_var_cov_mat"
## [7] "fit_dispersion" "fit_model_statistics" "fit_algorithm"
## [10] "fit_model_summary"In particular, we can see how fit_coeffs matches the results obtained in the UI (Figure 4.4):
fit_results$fit_coeffs## estimate std.error statistic p.value
## coeff_C 0.001280319 0.0004714055 2.715961 6.608367e-03
## coeff_alpha 0.021038724 0.0051576170 4.079156 4.519949e-05
## coeff_beta 0.063032534 0.0040073856 15.729091 9.557291e-56To visualise the dose-effect curve, we call the plot_fit_dose_curve() function:
plot_fit_dose_curve(
fit_results,
aberr_name = "Dicentrics",
place = "UI"
)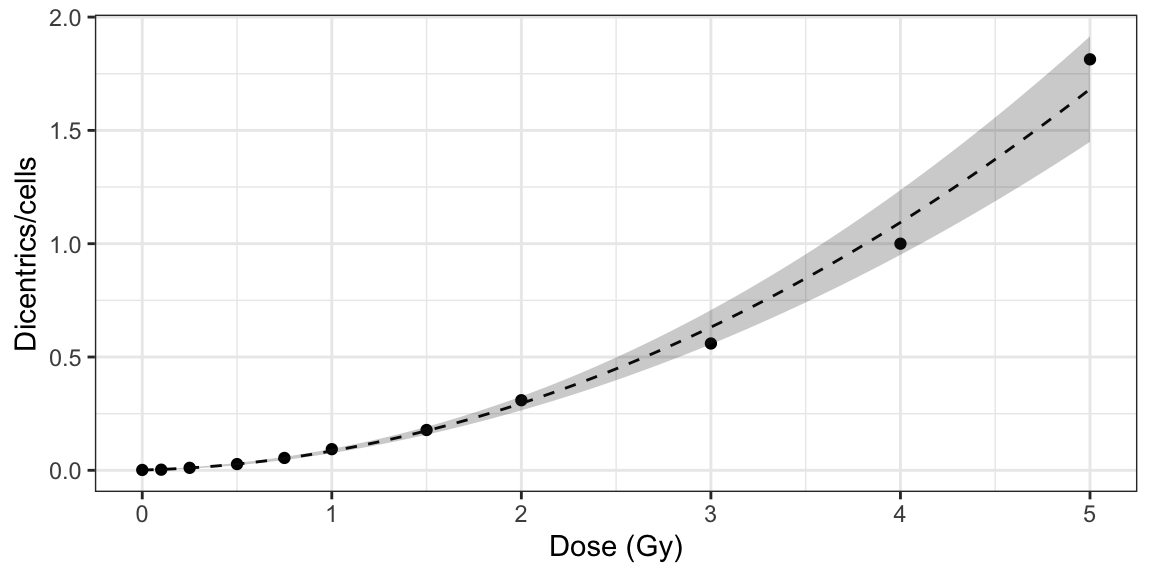
Figure 5.1: Plot of dose-effect curve generated by {biodosetools}. The grey shading indicates the uncertainties associated with the calibration curve.
5.2 Dicentrics dose estimation
5.2.1 Input case data
Next we can choose to either load the case data from a file or to input the data manually. The data is then parsed in R by calling the calculate_aberr_table() function. This will calculate the total number of cells (\(N\)), total number of aberrations (\(X\)), as well as mean (\(\bar{y}\)), standard error (\(\sigma\)), dispersion index (\(\sigma^{2}/\bar{y}\)), and \(u\)-value.
case_data <- system.file("extdata", "cases-data-partial.csv",
package = "biodosetools"
) |>
utils::read.csv(header = TRUE) |>
calculate_aberr_table(
type = "case",
assessment_u = 1,
aberr_module = "dicentrics"
)
case_data## # A tibble: 2 × 13
## ID N X C0 C1 C2 C3 C4 C5 y y_err DI u
## <chr> <int> <int> <int> <int> <int> <int> <int> <int> <dbl> <dbl> <dbl> <dbl>
## 1 exam… 361 100 302 28 22 8 1 0 0.277 0.0368 1.77 10.4
## 2 exam… 264 200 160 55 19 17 9 4 0.758 0.0736 1.89 10.25.2.2 Perform dose estimation
Finally, to perform the dose estimation in R we can call the adequate estimate_*() functions and parameters depending on the characteristics of the accident. In this example, we will use estimate_whole_body_merkle() and estimate_partial_body_dolphin(). First of all, however, we will need to load the fit coefficients and variance-covariance matrix from the previously calculated fit_results:
fit_coeffs <- fit_results$fit_coeffs
fit_var_cov_mat <- fit_results$fit_var_cov_matAfter that is done, we can simply call estimate_whole_body_merkle() and estimate_partial_body_dolphin():
results_whole_merkle <- estimate_whole_body_merkle(
num_cases = 1,
case_data,
fit_coeffs,
fit_var_cov_mat,
conf_int_yield = 0.83,
conf_int_curve = 0.83,
protracted_g_value = 1,
aberr_module = "dicentrics"
)
results_partial <- estimate_partial_body_dolphin(
num_cases = 1,
case_data,
fit_coeffs,
fit_var_cov_mat,
conf_int = 0.95,
gamma = 1 / 2.7,
aberr_module = "dicentrics"
)To visualise the estimated doses, we call the plot_estimated_dose_curve() function:
plot_estimated_dose_curve(
est_doses = list(
whole = results_whole_merkle,
partial = results_partial
),
fit_coeffs,
fit_var_cov_mat,
protracted_g_value = 1,
conf_int_curve = 0.95,
aberr_name = "Dicentrics",
place = "UI"
)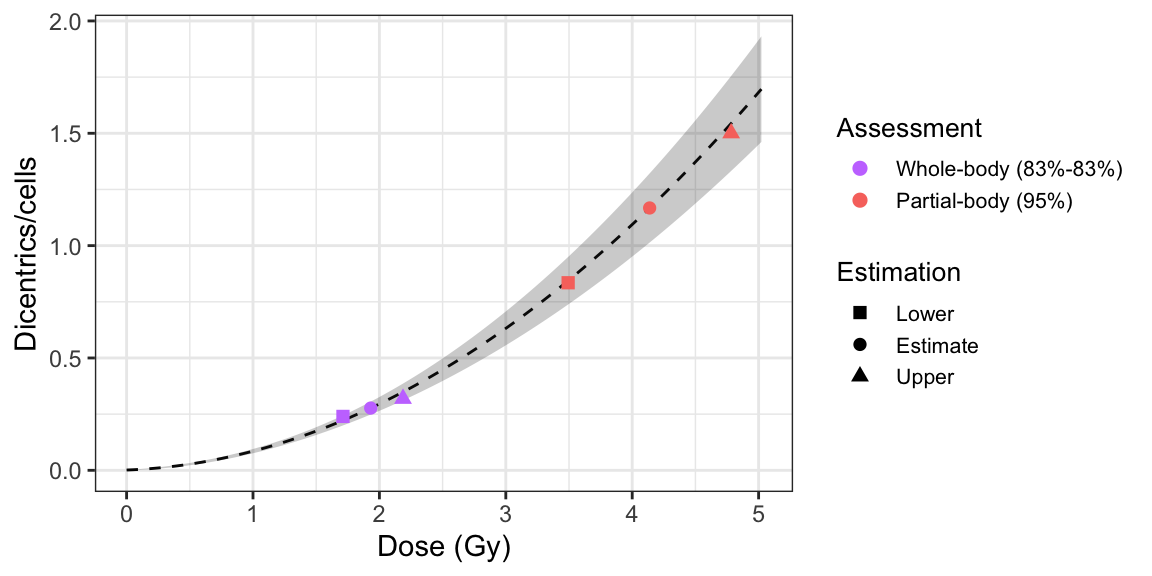
Figure 5.2: Plot of estimated doses generated by {biodosetools}. The grey shading indicates the uncertainties associated with the calibration curve.
5.3 Translocations dose-effect fitting
5.3.1 Calculate genomic conversion factor
To be able to fit the equivalent full genome dose-effect curve, we need to calculate the genomic conversion factor. To calculate the genomic conversion factor in R we call the calculate_genome_factor() function:
genome_factor <- calculate_genome_factor(
dna_table = dna_content_fractions_morton,
chromosome = c(1, 4, 11),
color = rep("Red", 3),
sex = "female"
)
genome_factor## [1] 0.31477975.3.2 Input count data
Once the genomic conversion factor has been calculated, we can input the count data. This step is accomplished in R by calling the calculate_aberr_table() function. This will calculate the total number of cells (\(N\)), total number of aberrations (\(X\)), as well as mean (\(\bar{y}\)), variance (\(\sigma^{2}\)), dispersion index (\(\sigma^{2}/\bar{y}\)), and \(u\)-value.
count_data <- system.file("extdata", "count-data-rodriguez-2004.csv", package = "biodosetools") |>
utils::read.csv() |>
calculate_aberr_table(type = "count") |>
dplyr::mutate(N = N * genome_factor)
count_data## # A tibble: 11 × 13
## D N X C0 C1 C2 C3 C4 C5 mean var DI
## <dbl> <dbl> <dbl> <int> <int> <int> <int> <int> <int> <dbl> <dbl> <dbl>
## 1 0 1371. 6 4350 6 0 0 0 0 0.00138 0.00138 0.999
## 2 0.1 1046. 15 3309 15 0 0 0 0 0.00451 0.00449 0.996
## 3 0.25 966. 18 3051 18 0 0 0 0 0.00587 0.00583 0.994
## 4 0.5 967. 33 3039 33 0 0 0 0 0.0107 0.0106 0.990
## 5 0.75 664. 40 2072 38 1 0 0 0 0.0189 0.0195 1.03
## 6 1 669. 50 2075 48 1 0 0 0 0.0235 0.0239 1.02
## 7 1.5 328. 56 990 50 3 0 0 0 0.0537 0.0566 1.05
## 8 2 226. 71 650 65 3 0 0 0 0.0989 0.0976 0.987
## 9 3 246. 157 649 108 23 1 0 0 0.201 0.227 1.13
## 10 4 125. 147 265 117 15 0 0 0 0.370 0.310 0.836
## 11 5 124. 180 246 122 23 4 0 0 0.456 0.426 0.936
## # ℹ 1 more variable: u <dbl>5.3.3 Perform fitting
To perform the fitting in R we call the fit() function, whilst selecting the appropriate aberr_module ("translocations" in this example), model_formula ("lin-quad" or "lin" for LQ or L models, respectively), and model_family ("automatic", "poisson", or "quasipoisson") parameters:
fit_results <- fit(
count_data = count_data,
model_formula = "lin-quad",
model_family = "automatic",
fit_link = "identity",
aberr_module = "translocations"
)The fit_results object is a list that contains all necessary information about the count data as well as options selected when performing the fitting. This is a vital step to ensure traceability and reproducibility. Below we can see its elements:
names(fit_results)## [1] "fit_raw_data" "fit_formula_raw" "fit_formula_tex"
## [4] "fit_coeffs" "fit_cor_mat" "fit_var_cov_mat"
## [7] "fit_dispersion" "fit_model_statistics" "fit_algorithm"
## [10] "fit_model_summary"In particular, we can see how fit_coeffs matches the results obtained in the UI (Figure 4.13):
fit_results$fit_coeffs## estimate std.error statistic p.value
## coeff_C 0.006560406 0.002052834 3.195780 1.269259e-02
## coeff_alpha 0.027197296 0.009922177 2.741061 2.540751e-02
## coeff_beta 0.057982322 0.004630926 12.520675 1.550057e-06To visualise the dose-effect curve, we call the plot_fit_dose_curve() function:
plot_fit_dose_curve(
fit_results,
aberr_name = "Translocations",
place = "UI"
)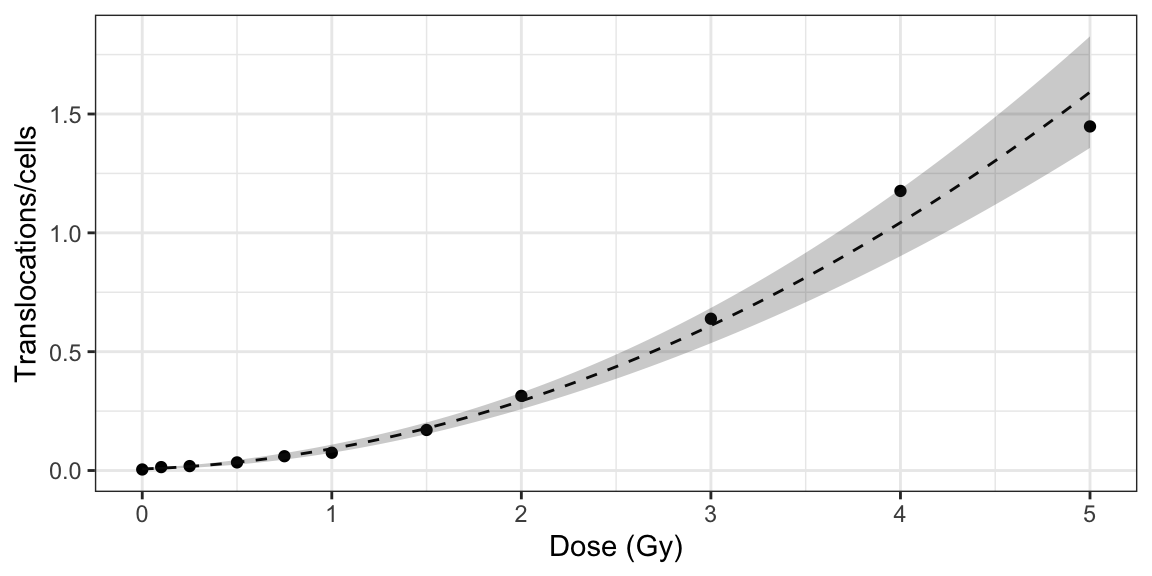
Figure 5.3: Plot of dose-effect curve generated by {biodosetools}. The grey shading indicates the uncertainties associated with the calibration curve.
5.4 Translocations dose estimation
5.4.1 Calculate genomic conversion factor
To be able to fit the equivalent full genome case data, we need to calculate the genomic conversion factor.
To do this, in the “Stain color options” box we select the sex of the individual, and the list of chromosomes and stains used for the translocation assay. Clicking on “Generate table” will show a table in the “Chromosome data” box in which we select the chromosome-stain pairs. Clicking on the “Calculate fraction” will calculate the genomic conversion factor.
To calculate the genomic conversion factor in R we call the calculate_genome_factor() function:
genome_factor <- calculate_genome_factor(
dna_table = dna_content_fractions_morton,
chromosome = c(1, 2, 3, 4, 5, 6),
color = c("Red", "Red", "Green", "Red", "Green", "Green"),
sex = "male"
)
genome_factor## [1] 0.58473435.4.2 Input case data
Next we can choose to either load the case data from a file or to input the data manually. The data is then parsed in R by calling the calculate_aberr_table() function. If needed, the user can select to use confounders (either using Sigurdson’s method, or by inputting the translocation frequency per cell). Once the table is generated and filled, the “Calculate parameters” button will calculate the total number of cells (\(N\)), total number of aberrations (\(X\)), as well as mean (\(\bar{F}_{p}\)), standard error (\(\sigma_{p}\)), dispersion index (\(\sigma^{2}/\bar{y}\)), \(u\)-value, expected translocation rate (\(X_{c}\)), full genome mean (\(\bar{F}_{g}\)), and full genome error (\(\sigma_{g}\)).
case_data <- data.frame(
ID = 'Case1', C0 = 288, C1 = 52, C2 = 9, C3 = 1
) |>
calculate_aberr_table(
type = "case",
assessment_u = 1,
aberr_module = "translocations"
) |>
dplyr::mutate(
Xc = calculate_trans_rate_sigurdson(
cells = N,
genome_factor = genome_factor,
age_value = 30,
smoker_bool = TRUE
),
Fg = (X - Xc) / (N * genome_factor),
Fg_err = Fp_err / sqrt(genome_factor)
)
case_data## # A tibble: 1 × 14
## ID N X C0 C1 C2 C3 Fp Fp_err DI u Xc Fg
## <chr> <int> <int> <int> <int> <int> <int> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 Case1 350 73 288 52 9 1 0.209 0.0259 1.12 1.64 1.02 0.352
## # ℹ 1 more variable: Fg_err <dbl>5.4.3 Perform dose estimation
Finally, to perform the dose estimation in R we can call the adequate estimate_*() functions and parameters depending on the characteristics of the accident. In this example, we will use estimate_whole_body_merkle(). First of all, however, we will need to load the fit coefficients and variance-covariance matrix from the previously calculated fit_results:
fit_coeffs <- fit_results$fit_coeffs
fit_var_cov_mat <- fit_results$fit_var_cov_matSince we have a protracted exposure, we need to calculate the value of \(G(x)\):
protracted_g_value <- protracted_g_function(
time = 0.5,
time_0 = 2
)
protracted_g_value## [1] 0.9216251After that is done, we can simply call estimate_whole_body_merkle():
results_whole_delta <- estimate_whole_body_merkle(
num_cases = 1,
case_data,
fit_coeffs,
fit_var_cov_mat,
conf_int_yield = 0.83,
conf_int_curve = 0.83,
protracted_g_value = 1,
genome_factor,
aberr_module = "translocations"
)To visualise the estimated doses, we call the plot_estimated_dose_curve() function:
plot_estimated_dose_curve(
est_doses = list(whole = results_whole_delta),
fit_coeffs,
fit_var_cov_mat,
protracted_g_value,
conf_int_curve = 0.95,
aberr_name = "Translocations",
place = "UI"
)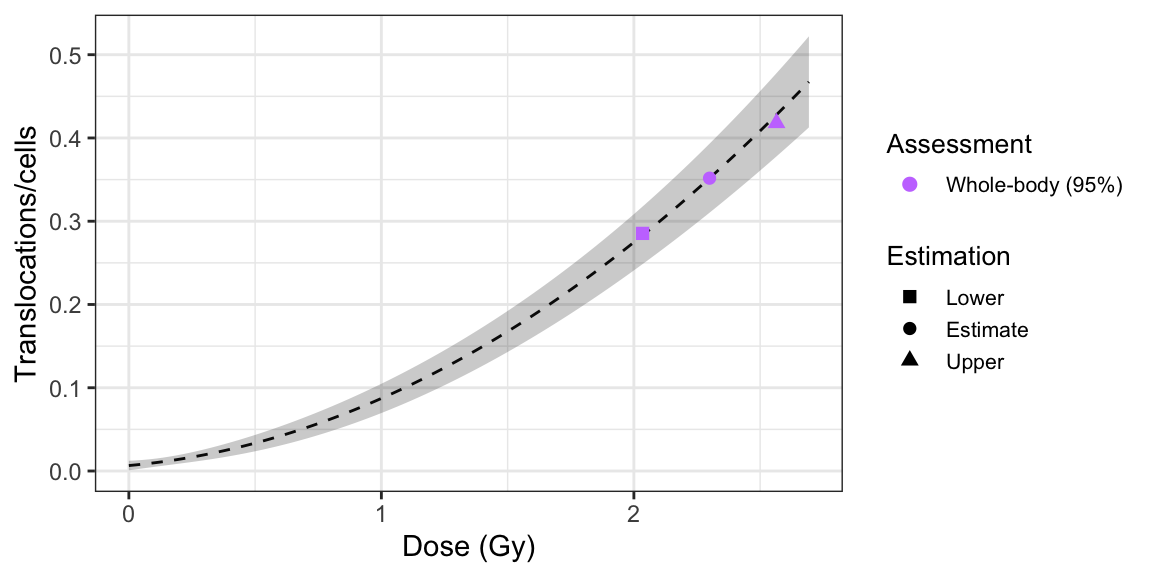
Figure 5.4: Plot of estimated doses generated by {biodosetools}. The grey shading indicates the uncertainties associated with the calibration curve.